SPACEc
A Stanford library for multiplexed imaging analysis, from cell segmentation through spatial analysis.
Python · six core modules · published on PyPI and bioRxiv
An interactive Python workflow for multiplexed image processing, from tissue extraction to spatial analysis, in one library.
sp.tl.cellpose_segmentation(img, ...)
sp.pp.filter_cells(adata, ...)
sp.tl.leiden(adata, resolution=1.0)
sp.tl.annotate(adata, method='stellar')
sp.tl.patch_proximity(adata, ...)
sp.pl.spatial(adata, color='cell_type')
The Problem
Multiplexed imaging lets researchers see 40+ proteins in a single tissue section, which matters for studying how cells interact in cancer, autoimmunity, and neuroscience. But the analysis is stitched together from separate tools: Cellpose for segmentation, scanpy for clustering, custom scripts for spatial analysis, matplotlib for visualization.
That means more time spent wrangling code than doing science. Each lab ends up with its own collection of notebooks that break when a dependency updates.
The Idea
One library that handles the whole workflow, from raw TIFF images to spatial results, behind a consistent API. SPACEc follows the scanpy convention: sp.tl (tools), sp.pp (preprocessing), sp.pl (plotting), sp.hf (helpers). It's built on AnnData, the standard format in single-cell biology.
Architecture
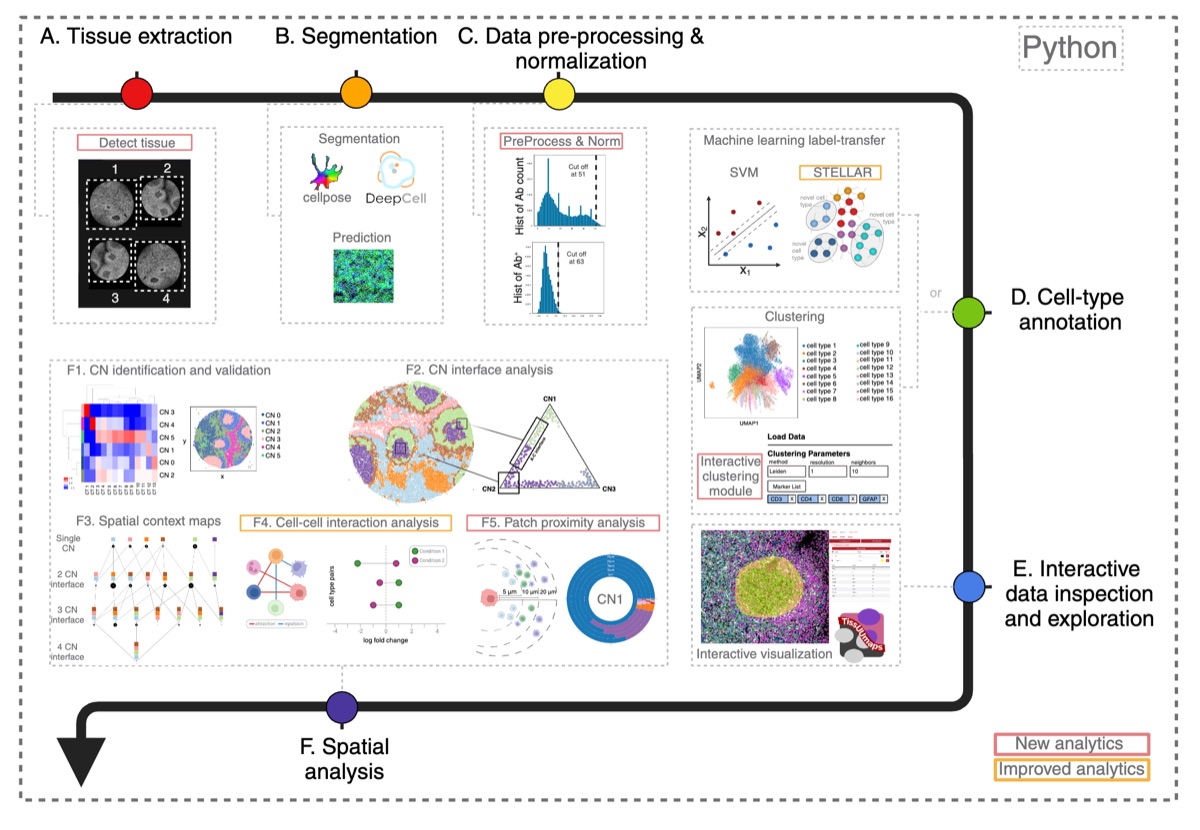
Full pipeline overview from the SPACEc paper
TIFF images
CSV markers
DeepCell (Mesmer)
QuPath import
Noise removal
Normalization
FlowSOM
GPU (RAPIDS)
STELLAR (GNN)
Hyperparameter tuning
Cell interactions
Patch Proximity*
*Patch Proximity Analysis is the novel research contribution.
Module Structure
- sp.tl._general — core analysis, clustering, annotation, spatial
- sp.pl._general — visualization and plotting
- sp.tl._segmentation — Cellpose, DeepCell, image processing
- sp.hf._general — helper functions
- sp.pp._general — preprocessing pipelines
- sp._shared.segmentation — shared segmentation logic
The Novel Contribution
Patch Proximity Analysis
Standard neighborhood analysis works on cell-to-cell distances. It tells you cell A is near cell B, but not how groups of cells are organized at the tissue level.
Patch Proximity Analysis takes a different route. DBSCAN finds spatial clusters of a given cell type (patches of tumor cells, immune aggregates, stromal clusters), concave hulls draw boundaries around each patch, and the analysis then measures how other cell types sit relative to those boundaries: inside, on the border, or nearby.
That picks up tissue-level organization the cell-level view misses. A tumor microenvironment isn't just a set of pairwise interactions; it's the arrangement of which populations sit next to which.
Decisions
AnnData
AnnData is the standard data structure in single-cell biology, so building on it lets SPACEc work directly with scanpy. Researchers don't learn a new format, and they can move between the two. The .obs, .var, and .obsm fields map cleanly onto cell metadata, marker metadata, and spatial coordinates.
Both Cellpose and DeepCell
Different imaging modalities call for different segmentation. Cellpose does well on cytoplasm with limited training data; DeepCell (Mesmer) is better for nuclear segmentation in whole-cell multiplex images. Supporting both lets researchers pick the right tool without leaving the library.
GPU acceleration
These datasets get large: tens of thousands of cells per sample, hundreds of samples per study. Leiden clustering that takes about 20 minutes on CPU runs in under 2 on GPU via RAPIDS, which matters when you're iterating.
The scanpy API convention
Most computational biologists already know sc.tl, sc.pp, and sc.pl. Mirroring it with sp.tl, sp.pp, and sp.pl keeps the learning curve on the science rather than the API, which makes people more likely to actually use the library.
Tradeoffs
Python 3.9–3.10 only. TensorFlow, PyTorch, and RAPIDS have version requirements that don't overlap cleanly above 3.10. This is the biggest friction point for adoption, which is why the CPU and GPU Docker images are the recommended install path.
Monolithic modules. The _general.py files are large and hard to navigate. The scanpy convention keeps the public API consistent (sp.tl.function()) at the cost of file organization. That was a deliberate call: clean API, dense internals.
Dependency sprawl. Cellpose, DeepCell, RAPIDS, PyTorch Geometric, GeoPandas, and TensorFlow each pull in transitive dependencies that fight each other, so a clean install outside Docker takes real effort. That's the cost of wrapping the best tool for each step instead of reimplementing them.
Talk
